Create a subtree with largest overlap from a species list.
Source:R/phylo_builder.R
phylobuilder.Rdphylobuilder creates a subtree with largest overlap from a species list. If species in the species list are not already in the tip label, species will be added at the most recent common ancestor at the genus or family level when possible.
phylobuilder(species, tree, extract = TRUE)Arguments
- species
A vector or matrix containing a species list
- tree
a phylogenetic tree (object of class phylo)
- extract
extract the species in the list after trying to add missing labels to the tree. If FALSE phylobuilder adds only the taxa in the list.
Value
phylobuilder returns a phylogenetic tree, i.e. an object of
class phylo.
See also
Examples
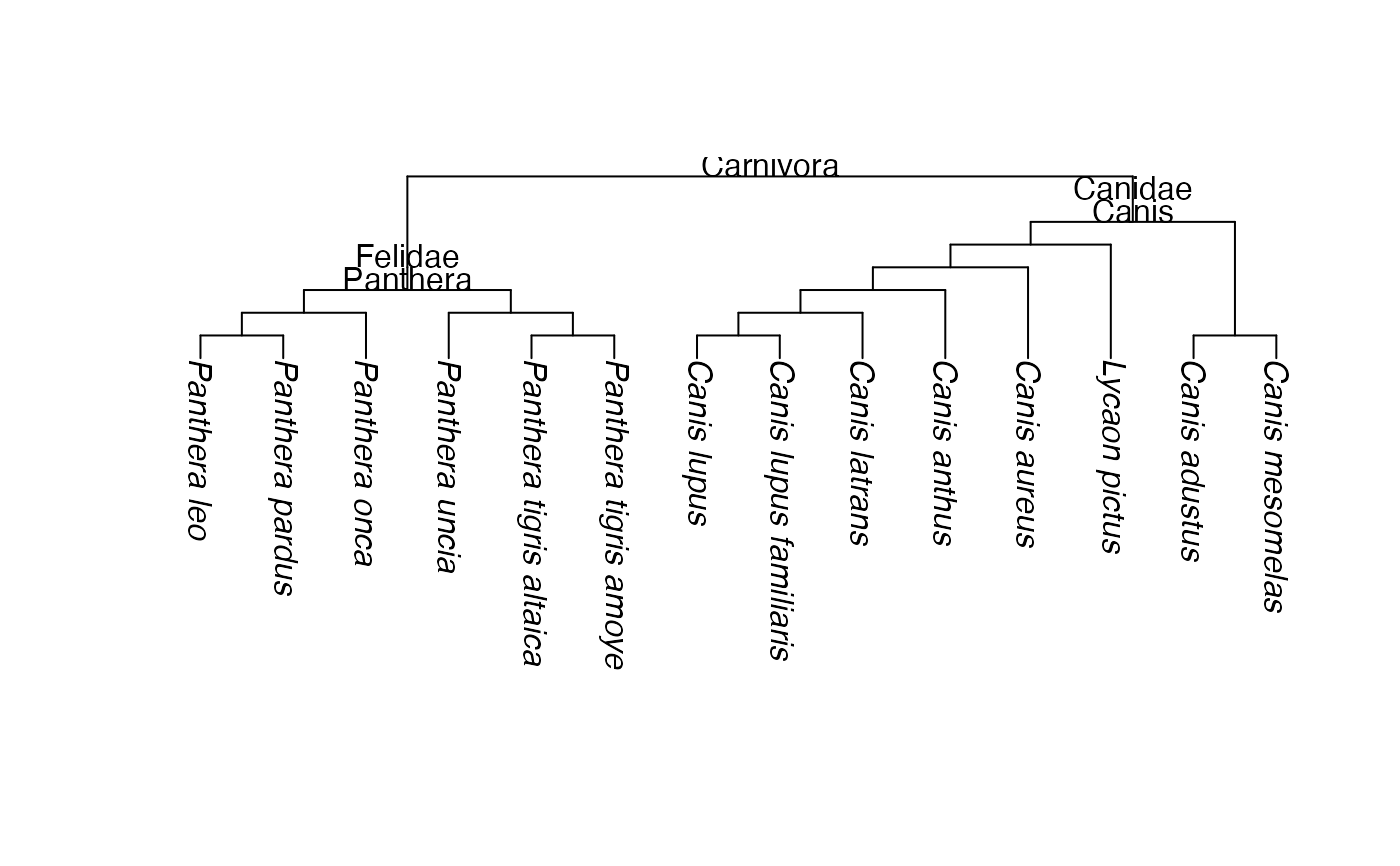
library(ape)
txt <- "(((((Panthera_leo,Panthera_pardus), Panthera_onca),(Panthera_uncia,
(Panthera_tigris_altaica, Panthera_tigris_amoyensis)))Panthera)Felidae,
(((((((Canis_lupus,Canis_lupus_familiaris),Canis_latrans),Canis_anthus),
Canis_aureus),Lycaon_pictus),(Canis_adustus,Canis_mesomelas))Canis)
Canidae)Carnivora;"
txt <- gsub("[[:space:]]", "", txt)
cats_and_dogs <- read.tree(text=txt)
plot(cats_and_dogs, node.depth=2, direction="downwards")
nodelabels(cats_and_dogs$node.label, frame="none", adj = c(0.5, 0))
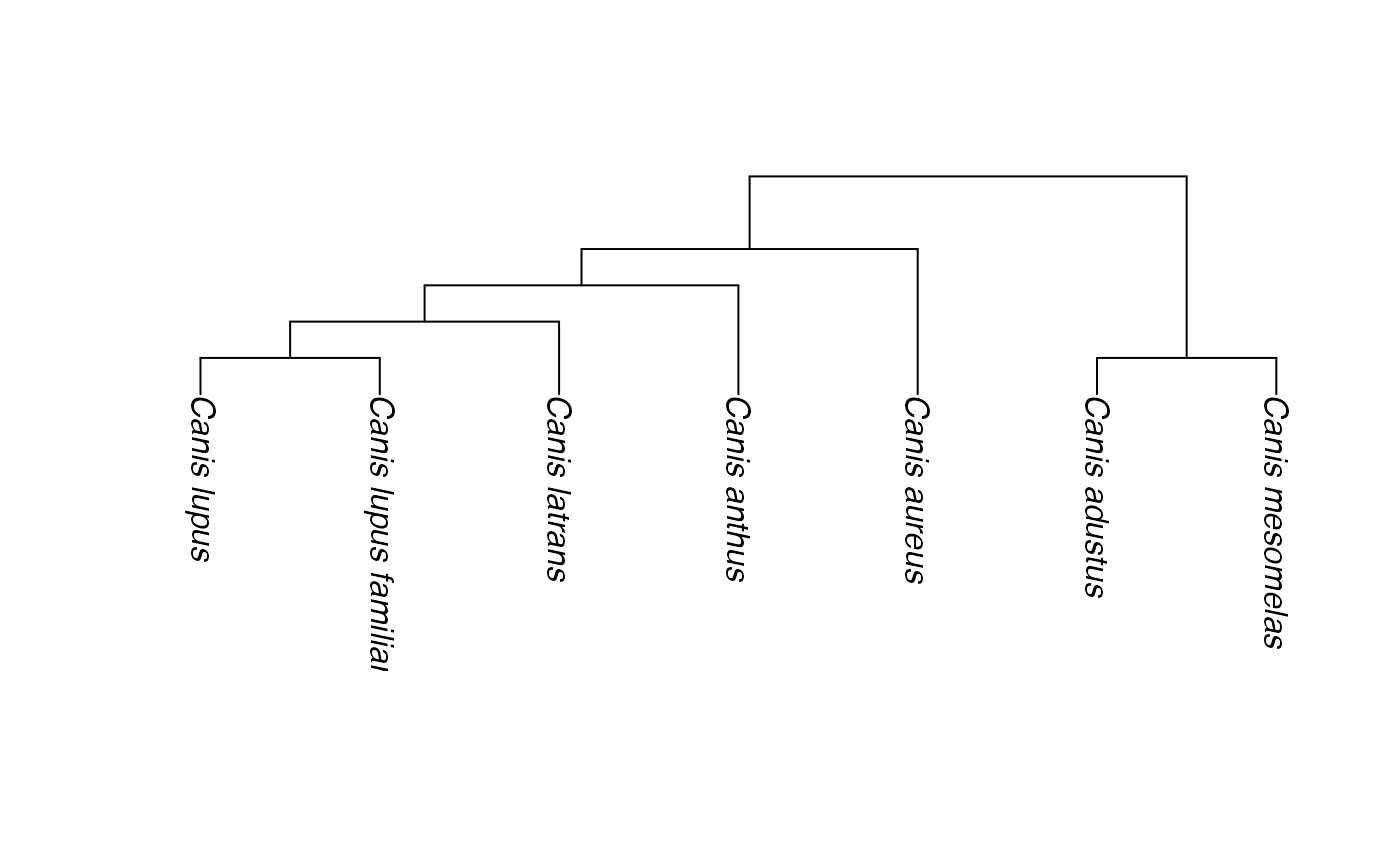
 tree <- drop.tip(cats_and_dogs, c("Panthera_uncia", "Lycaon_pictus"),
collapse.singles=FALSE)
dogs <- c("Canis_lupus", "Canis_lupus_familiaris", "Canis_latrans",
"Canis_anthus", "Canis_aureus", "Lycaon_pictus", "Canis_adustus",
"Canis_mesomelas")
# try to extract tree with all 'dogs'
t1 <- phylobuilder(dogs, tree)
plot(t1, direction="downwards")
tree <- drop.tip(cats_and_dogs, c("Panthera_uncia", "Lycaon_pictus"),
collapse.singles=FALSE)
dogs <- c("Canis_lupus", "Canis_lupus_familiaris", "Canis_latrans",
"Canis_anthus", "Canis_aureus", "Lycaon_pictus", "Canis_adustus",
"Canis_mesomelas")
# try to extract tree with all 'dogs'
t1 <- phylobuilder(dogs, tree)
plot(t1, direction="downwards")
 attr(t1, "species_list")
#> species added
#> [1,] "Canis_lupus" "tree"
#> [2,] "Canis_lupus_familiaris" "tree"
#> [3,] "Canis_latrans" "tree"
#> [4,] "Canis_anthus" "tree"
#> [5,] "Canis_aureus" "tree"
#> [6,] "Lycaon_pictus" "missing"
#> [7,] "Canis_adustus" "tree"
#> [8,] "Canis_mesomelas" "tree"
# providing extra information ("Family", "Order", ...) can help
sp <- data.frame(Order = c("Carnivora", "Carnivora", "Carnivora"),
Family = c("Felidae", "Canidae", "Canidae"),
Genus = c("Panthera", "Lycaon", "Vulpes"),
Species = c("uncia", "pictus", "vulpes"),
Common_name = c("Snow leopard", "Africa wild dog", "Red fox"))
sp
#> Order Family Genus Species Common_name
#> 1 Carnivora Felidae Panthera uncia Snow leopard
#> 2 Carnivora Canidae Lycaon pictus Africa wild dog
#> 3 Carnivora Canidae Vulpes vulpes Red fox
# Now we just add some species
t2 <- phylobuilder(sp, tree, extract=FALSE)
plot(t2, direction="downwards")
attr(t1, "species_list")
#> species added
#> [1,] "Canis_lupus" "tree"
#> [2,] "Canis_lupus_familiaris" "tree"
#> [3,] "Canis_latrans" "tree"
#> [4,] "Canis_anthus" "tree"
#> [5,] "Canis_aureus" "tree"
#> [6,] "Lycaon_pictus" "missing"
#> [7,] "Canis_adustus" "tree"
#> [8,] "Canis_mesomelas" "tree"
# providing extra information ("Family", "Order", ...) can help
sp <- data.frame(Order = c("Carnivora", "Carnivora", "Carnivora"),
Family = c("Felidae", "Canidae", "Canidae"),
Genus = c("Panthera", "Lycaon", "Vulpes"),
Species = c("uncia", "pictus", "vulpes"),
Common_name = c("Snow leopard", "Africa wild dog", "Red fox"))
sp
#> Order Family Genus Species Common_name
#> 1 Carnivora Felidae Panthera uncia Snow leopard
#> 2 Carnivora Canidae Lycaon pictus Africa wild dog
#> 3 Carnivora Canidae Vulpes vulpes Red fox
# Now we just add some species
t2 <- phylobuilder(sp, tree, extract=FALSE)
plot(t2, direction="downwards")
 attr(t2, "species_list")
#> species added
#> [1,] "Panthera_uncia" "genus"
#> [2,] "Lycaon_pictus" "family"
#> [3,] "Vulpes_vulpes" "family"
attr(t2, "species_list")
#> species added
#> [1,] "Panthera_uncia" "genus"
#> [2,] "Lycaon_pictus" "family"
#> [3,] "Vulpes_vulpes" "family"