Benchmarking phyloregion
Barnabas H. Daru, Piyal Karunarathne & Klaus Schliep
November 15, 2022
Source:vignettes/Benchmark.Rmd
Benchmark.RmdBenchmarking phyloregion with comparable packages
In this vignette, we benchmark phyloregion against other
similar R packages in analyses of standard alpha diversity
metrics commonly used in conservation, such as phylogenetic diversity
and phylogenetic endemism as well as metrics for analyzing compositional
turnover (e.g., beta diversity and phylogenetic beta diversity).
Specifically, we compare phyloregion’s functions with
available packages for efficiency in memory allocation and computation
speed in various biogeographic analyses.
First, load the packages for the benchmarking:
library(ape)
library(Matrix)
library(bench)
library(ggplot2)
# packages we benchmark
library(phyloregion)
library(betapart)
library(picante)
library(vegan)
library(hilldiv)
library(BAT)
library(pez)We will use a small data set which comes with
phyloregion. This dataset consists of a dated phylogeny of
the woody plant species of southern Africa along with their geographical
distributions. The dataset comes from a study that maps tree diversity
hotspots in southern Africa (Daru, Bank, and
Davies 2015).
data(africa)
# subset matrix
X_sparse <- africa$comm[1:30, ]
X_sparse <- X_sparse[, colSums(X_sparse)>0]
X_dense <- as.matrix(X_sparse)
Xt_dense <- t(X_dense)
object.size(X_sparse)## 76504 bytes
object.size(X_dense)## 134752 bytes
dim(X_sparse)## [1] 30 401To make results comparable, it is often desirable to make sure that
the taxa in different datasets match each other (Kembel et al. 2010). For example, the community
matrix in the hilldiv package (Alberdi 2019) needs to be transposed. These
transformations can influence the execution times of the function, often
only marginally. Thus, to benchmark phyloregion against
other packages, we here use the package bench (Hester 2020) because it returns execution times
and provides estimates of memory allocations for each computation.
1. Analysis of alpha diversity
1.1. Benchmarking phyloregion for analysis of
phylogenetic diversity
For analysis of alpha diversity commonly used in conservation such as
phylogenetic diversity - the sum of all phylogenetic branch lengths
within an area (Faith 1992) -
phyloregion is 31 to 284 times faster and 67 to 192 times
memory efficient, compared to other packages!
tree <- africa$phylo
tree <- keep.tip(tree, colnames(X_sparse))
pd_picante <- function(x, tree){
res <- picante::pd(x, tree)[,1]
names(res) <- row.names(x)
res
}
pd_pez <- function(x, tree){
dat <- pez::comparative.comm(tree, x)
res <- pez::.pd(dat)[,1]
names(res) <- row.names(x)
res
}
pd_hilldiv <- function(x, tree) hilldiv::index_div(x, tree, index="faith")
pd_phyloregion <- function(x, tree) phyloregion::PD(x, tree)
res1 <- bench::mark(picante=pd_picante(X_dense, tree),
hilldiv=pd_hilldiv(Xt_dense,tree=tree),
pez=pd_pez(X_dense, tree),
phyloregion=pd_phyloregion(X_sparse, tree))
summary(res1)## # A tibble: 4 × 6
## expression min median `itr/sec` mem_alloc `gc/sec`
## <bch:expr> <bch:tm> <bch:tm> <dbl> <bch:byt> <dbl>
## 1 picante 97.85ms 112.45ms 8.22 59.6MB 9.87
## 2 hilldiv 762.42ms 762.42ms 1.31 170.1MB 3.93
## 3 pez 108.59ms 122.83ms 8.16 60.4MB 8.16
## 4 phyloregion 2.82ms 3.23ms 268. 909.8KB 5.99
autoplot(res1)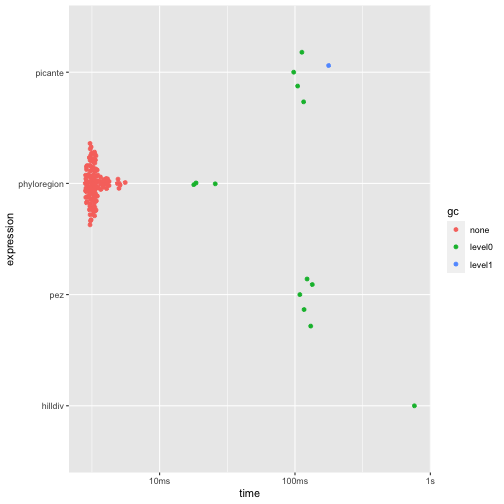
1.2. Benchmarking phyloregion for analysis of
phylogenetic endemism
Another benchmark for phyloregion is in the analysis of
phylogenetic endemism, the degree to which phylogenetic diversity is
restricted to any given area (Rosauer et al.
2009). Here, we found that phyloregion is 160 times
faster and 489 times efficient in memory allocation.
tree <- africa$phylo
tree <- keep.tip(tree, colnames(X_sparse))
pe_pez <- function(x, tree){
dat <- pez::comparative.comm(tree, x)
res <- pez::pez.endemism(dat)[,1]
names(res) <- row.names(x)
res
}
pe_phyloregion <- function(x, tree) phyloregion::phylo_endemism(x, tree)
res2 <- bench::mark(pez=pe_pez(X_dense, tree),
phyloregion=pe_phyloregion(X_sparse, tree))
summary(res2)## # A tibble: 2 × 6
## expression min median `itr/sec` mem_alloc `gc/sec`
## <bch:expr> <bch:tm> <bch:tm> <dbl> <bch:byt> <dbl>
## 1 pez 630.38ms 630.38ms 1.59 493.88MB 9.52
## 2 phyloregion 2.94ms 3.19ms 280. 1.06MB 3.98
autoplot(res2)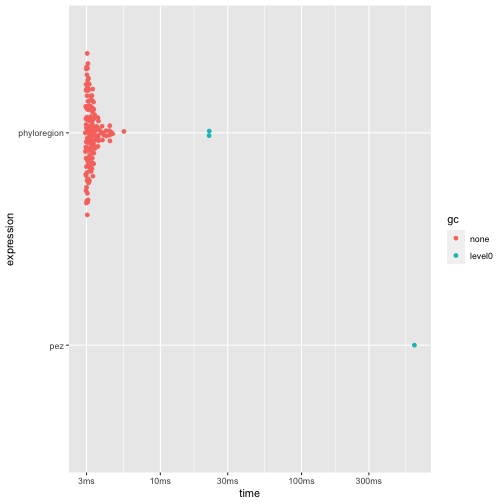
2. Analysis of compositional turnover (beta diversity)
2.1. Benchmarking phyloregion for analysis of taxonomic
beta diversity
For analysis of taxonomic beta diversity, which compares diversity
between communities (Koleff, Gaston, and Lennon
2003), phyloregion has marginal advantage over other
packages. Nonetheless, it is 1-39 times faster and allocates 2 to 110
times less memory than other packages.
chk_fun <- function(target, current)
all.equal(target, current, check.attributes = FALSE)
fun_phyloregion <- function(x) as.matrix(phyloregion::beta_diss(x)[[3]])
fun_betapart <- function(x) as.matrix(betapart::beta.pair(x)[[3]])
fun_vegan <- function(x) as.matrix(vegan::vegdist(x, binary=TRUE))
fun_BAT <- function(x) as.matrix(BAT::beta(x, func = "Soerensen")[[1]])
res3 <- bench::mark(phyloregion=fun_phyloregion(X_sparse),
betapart=fun_betapart(X_dense),
vegan=fun_vegan(X_dense),
BAT=fun_BAT(X_dense), check=chk_fun)
summary(res3)## # A tibble: 4 × 6
## expression min median `itr/sec` mem_alloc `gc/sec`
## <bch:expr> <bch:tm> <bch:tm> <dbl> <bch:byt> <dbl>
## 1 phyloregion 598.5µs 689µs 1210. 428.2KB 4.39
## 2 betapart 849.4µs 909µs 1024. 594.1KB 10.3
## 3 vegan 946.8µs 992µs 958. 1016.1KB 7.01
## 4 BAT 44.5ms 47ms 20.9 31.8MB 8.95
autoplot(res3)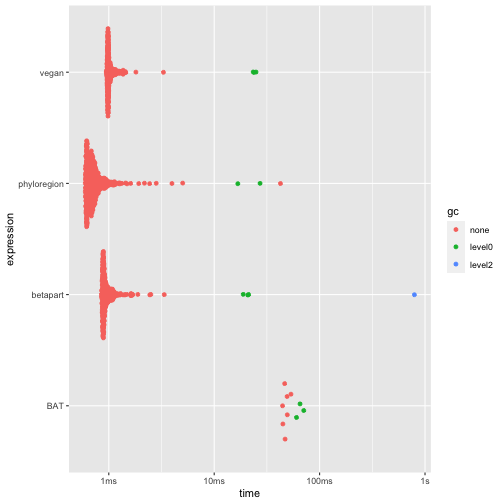
2.2. Benchmarking phyloregion for analysis of
phylogenetic beta diversity
For analysis of phylogenetic turnover (beta-diversity) among
communities - the proportion of shared phylogenetic branch lengths
between communities (Graham and Fine 2008)
- phyloregion is 300-400 times faster and allocates 100-600
times less memory!
fun_phyloregion <- function(x, tree) phyloregion::phylobeta(x, tree)[[3]]
fun_betapart <- function(x, tree) betapart::phylo.beta.pair(x, tree)[[3]]
fun_picante <- function(x, tree) 1 - picante::phylosor(x, tree)
fun_BAT <- function(x, tree) BAT::beta(x, tree, func = "Soerensen")[[1]]
chk_fun <- function(target, current)
all.equal(target, current, check.attributes = FALSE)
res4 <- bench::mark(picante=fun_picante(X_dense, tree),
betapart=fun_betapart(X_dense, tree),
BAT=fun_BAT(X_dense, tree),
phyloregion=fun_phyloregion(X_sparse, tree), check=chk_fun)
summary(res4)## # A tibble: 4 × 6
## expression min median `itr/sec` mem_alloc `gc/sec`
## <bch:expr> <bch:tm> <bch:tm> <dbl> <bch:byt> <dbl>
## 1 picante 2.25s 2.25s 0.444 1.24GB 2.66
## 2 betapart 2.05s 2.05s 0.489 1.24GB 2.93
## 3 BAT 1.41s 1.41s 0.712 293.64MB 0.712
## 4 phyloregion 4.21ms 4.49ms 175. 1.12MB 1.99
autoplot(res4)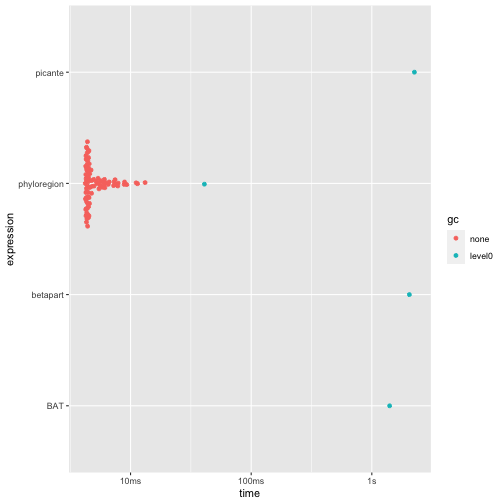
Note that for this test, picante returns a similarity
matrix while betapart, and phyloregion return
a dissimilarity matrix.
Session Information
## R version 4.2.1 (2022-06-23)
## Platform: x86_64-apple-darwin17.0 (64-bit)
## Running under: macOS Monterey 12.6
##
## Matrix products: default
## LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] pez_1.2-4 BAT_2.9.2 hilldiv_1.5.1 picante_1.8.2
## [5] nlme_3.1-157 vegan_2.6-2 lattice_0.20-45 permute_0.9-7
## [9] betapart_1.5.6 phyloregion_1.0.7 bench_1.1.2 Matrix_1.5-3
## [13] ape_5.6-2 knitr_1.39 ggplot2_3.3.6
##
## loaded via a namespace (and not attached):
## [1] utf8_1.2.2 proto_1.0.0 ks_1.13.5
## [4] tidyselect_1.2.0 htmlwidgets_1.5.4 grid_4.2.1
## [7] combinat_0.0-8 pROC_1.18.0 animation_2.7
## [10] munsell_0.5.0 codetools_0.2-18 clustMixType_0.2-15
## [13] interp_1.1-3 future_1.29.0 withr_2.5.0
## [16] profmem_0.6.0 colorspace_2.0-3 highr_0.9
## [19] rstudioapi_0.13 geometry_0.4.6.1 stats4_4.2.1
## [22] ggsignif_0.6.4 listenv_0.8.0 slam_0.1-50
## [25] FD_1.0-12.1 nls2_0.3-3 mnormt_2.1.0
## [28] farver_2.1.1 coda_0.19-4 parallelly_1.32.1
## [31] vctrs_0.5.0 generics_0.1.3 clusterGeneration_1.3.7
## [34] ipred_0.9-13 xfun_0.31 itertools_0.1-3
## [37] fastcluster_1.2.3 R6_2.5.1 doParallel_1.0.17
## [40] ggbeeswarm_0.6.0 pdist_1.2.1 assertthat_0.2.1
## [43] scales_1.2.0 nnet_7.3-17 beeswarm_0.4.0
## [46] rgeos_0.5-9 gtable_0.3.0 globals_0.16.1
## [49] caper_1.0.1 phangorn_2.9.0 MatrixModels_0.5-1
## [52] timeDate_4021.106 rlang_1.0.6 FSA_0.9.3
## [55] scatterplot3d_0.3-41 splines_4.2.1 rstatix_0.7.1
## [58] rgdal_1.5-30 ModelMetrics_1.2.2.2 broom_1.0.1
## [61] checkmate_2.1.0 reshape2_1.4.4 abind_1.4-5
## [64] backports_1.4.1 Hmisc_4.7-1 caret_6.0-93
## [67] tools_4.2.1 lava_1.7.0 psych_2.2.9
## [70] lavaan_0.6-12 ellipsis_0.3.2 raster_3.5-21
## [73] RColorBrewer_1.1-3 proxy_0.4-27 Rcpp_1.0.9
## [76] plyr_1.8.7 base64enc_0.1-3 progress_1.2.2
## [79] purrr_0.3.4 prettyunits_1.1.1 ggpubr_0.4.0
## [82] rpart_4.1.16 deldir_1.0-6 pbapply_1.5-0
## [85] deSolve_1.34 qgraph_1.9.2 cluster_2.1.3
## [88] magrittr_2.0.3 data.table_1.14.2 SparseM_1.81
## [91] mvtnorm_1.1-3 smoothr_0.2.2 hms_1.1.1
## [94] evaluate_0.15 jpeg_0.1-9 mclust_6.0.0
## [97] gridExtra_2.3 compiler_4.2.1 tibble_3.1.8
## [100] maps_3.4.0 KernSmooth_2.23-20 crayon_1.5.1
## [103] hypervolume_3.0.4 htmltools_0.5.3 mgcv_1.8-40
## [106] corpcor_1.6.10 Formula_1.2-4 snow_0.4-4
## [109] tidyr_1.2.0 expm_0.999-6 lubridate_1.8.0
## [112] DBI_1.1.3 magic_1.6-0 subplex_1.8
## [115] MASS_7.3-57 ade4_1.7-19 car_3.1-1
## [118] cli_3.4.1 quadprog_1.5-8 parallel_4.2.1
## [121] gower_1.0.0 igraph_1.3.4 pkgconfig_2.0.3
## [124] numDeriv_2016.8-1.1 foreign_0.8-82 sp_1.5-0
## [127] terra_1.5-21 recipes_1.0.3 foreach_1.5.2
## [130] pbivnorm_0.6.0 predicts_0.1-3 vipor_0.4.5
## [133] hardhat_1.2.0 prodlim_2019.11.13 stringr_1.4.0
## [136] digest_0.6.29 pracma_2.4.2 phytools_1.0-3
## [139] rcdd_1.5 fastmatch_1.1-3 htmlTable_2.4.1
## [142] quantreg_5.94 gtools_3.9.3 geiger_2.0.10
## [145] lifecycle_1.0.3 glasso_1.11 carData_3.0-5
## [148] maptpx_1.9-7 fansi_1.0.3 pillar_1.8.0
## [151] fastmap_1.1.0 plotrix_3.8-2 survival_3.3-1
## [154] glue_1.6.2 fdrtool_1.2.17 png_0.1-7
## [157] iterators_1.0.14 class_7.3-20 stringi_1.7.8
## [160] palmerpenguins_0.1.1 doSNOW_1.0.20 latticeExtra_0.6-30
## [163] dplyr_1.0.9 e1071_1.7-11 future.apply_1.10.0